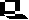
Picture to the left: Column chromatography. Separation of a mixture of three substances. The separation is based on the differences in the molecular retention due to the packing of the columnar material. Depending on the material, it is distinguished between ion exchange chromatography, molecular sieve chromatography, absorption chromatography etc. The material that has flown through a column is called eluate.
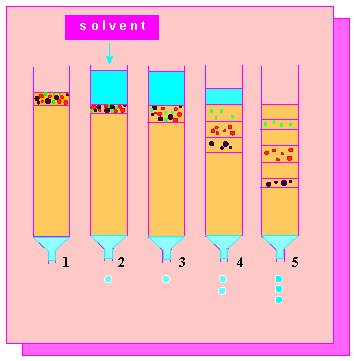
Picture to the right: Additional devices necessary for automatic columnar chromatographies: Scheme of a flow chart diagram.
The difficulty to obtain a certain molecule in a pure state is caused by the fact that all proteins are composed of the same units and that they have often more similarities than differences. There exist at least three ways to concentrate, isolate and characterize specific proteins. Usually, a combination of several subsequent purification steps is necessary.
![]() precipitation,
precipitation,
![]() chromatographic
separation,
chromatographic
separation,
![]() separation
in an electric field.
separation
in an electric field.
The first step is always the breaking off of the cells. Since this mixes plasma and vacuole contents, special precautions are necessary to avoid the denaturation of the proteins. The pH has to be kept stable by a suitable buffer and certain additions are necessary to neutralize toxic substances set free or unwanted enzyme activities.
Precipitation, for example by concentrated ammonia sulfate or by an organic solvent like acetone is often the first purification and concentration step. The precipitated material is centrifuged (sometimes requiring a high speed centrifuge, a so-called ultra centrifuge) and subsequently dissolved in a suitable solvent. What is suitable depends on the respective protein and the source material. Information about this can be found in course instructions or laboratory manuals, it is also given in the 'materials and methods' section of scientific publications. A chromatographic separation may be based on:
![]() different
sizes of the molecules (molecular sieve effect),
different
sizes of the molecules (molecular sieve effect),
![]() different
charges (different amounts of ionized groups within the
molecules),
different
charges (different amounts of ionized groups within the
molecules),
![]() or
a different affinity for the absorbing material.
or
a different affinity for the absorbing material.
One of the most common methods is column chromatography.
Paper chromatography has formerly been very common though it is used only rarely nowadays.
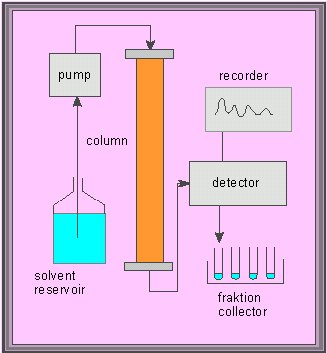
Scheme of a two-dimensional paper chromatogram. The paper (or any other carrier) is dried after finishing of the first run and turned by 90�. Two-dimensional chromatography exploits the different abilities to dissolve in different solvents (usually mixtures of several solvents)
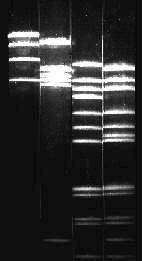 Besides
chromatography, electrophoresis plays an important part in all
analytical laboratories. Here, too, numerous variations exist. Most
common are separations by charge or by molecular size. The latter is
insofar problematic as the protein has to be denatured so that no
enzyme activity can be measured. This method plays an outstanding
part in the sequencing of DNA. It enables the separation of DNA
molecules with just one nucleotide difference in length in an
electric field. Normally, the material, like, for example, a mixture
of different proteins, is separated in a gel (polyacrylamide proved
to be very suitable); but it can also be worked with paper or starch
gels.
Besides
chromatography, electrophoresis plays an important part in all
analytical laboratories. Here, too, numerous variations exist. Most
common are separations by charge or by molecular size. The latter is
insofar problematic as the protein has to be denatured so that no
enzyme activity can be measured. This method plays an outstanding
part in the sequencing of DNA. It enables the separation of DNA
molecules with just one nucleotide difference in length in an
electric field. Normally, the material, like, for example, a mixture
of different proteins, is separated in a gel (polyacrylamide proved
to be very suitable); but it can also be worked with paper or starch
gels.
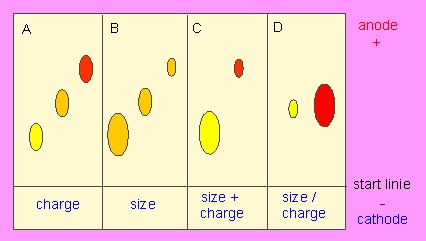

Principle of gel electrophoresis. Influence of charge and particle size on the electrophoretic mobility of proteins or other macromolecules like nucleic acids. A. Separation by charge, B. Separation by particle size, C: Addition of A and B, D: Compensation of A and B.
Isoelectric focusing is a modification of gel electrophoresis. It uses both an electric field and a pH-gradient so that the protein stops near its isoelectric point (pK). The isoelectric point is the pH where the smallest amount of ionized groups is present in the molecule. In an acidic field, -NH3+ and -COOH can be found, in the alkaline field are -NH2 and -COO- present and only at the isoelectric point exist both groups in their uncharged form (-NH2 and -COOH). It is therefore above all dependent on the ratio of the basic towards the acidic amino acids of a protein where it has its isoelectric point. Here, the solubility of the proteins is drastically reduced.
To define the purity of a protein, different criteria can be used:
![]() the
ability to crystallize,
the
ability to crystallize,
![]() the
solubility kinetics,
the
solubility kinetics,
![]() the
uniform behaviour in electrophoresis (just one band),
the
uniform behaviour in electrophoresis (just one band),
![]() clear
results when determining the amino acid sequence,
clear
results when determining the amino acid sequence,
![]() a
uniform catalytic activity (no side activities),
a
uniform catalytic activity (no side activities),
![]() uniformity
in the chromatographic behaviour in different solute systems.
uniformity
in the chromatographic behaviour in different solute systems.
It is always necessary to confirm several of these criteria independently of each other since none of them is sufficient enough by itself to make an irrefutable statement. Some proteins form crystals only in complex with other substances, others again have two different enzyme activities on just one polypeptide chain or the activity is dependent on a complex (a quarternary structure) of several maybe different polypeptide chains. A further group finally has an activity that depends on cofactors. Many of the membrane proteins are only active in connection with lipids, etc. The purity of a polypeptide chain and the enzyme activity are consequently two different quantities. It has therefore to be decided before starting an experiment what is to be achieved by a purification and where the gained material is needed for. Parallel attempts and working with different strategies belong to the every day life in a laboratory.
The demands have risen steadily. Today, it is often not enough any more to isolate a cell protein. The interest is much more to gain a protein of a specific compartment (cell nucleus, plastids, mitochondria, etc.) or to identify it as the component of a certain compartment. The purification of the protein is therefore preceeded by the concentration of the compartment. An important help is the density gradient centrifugation with an ultracentrifuge.
The sensitivity of the detection methods can be increased by ranges by the specific use of radioactively labelled source material.
|
|