Derivation of Thermodynamic Equations |
For an ideal gas undergoing a reversible change at constant temperature, when no chemical work is being performed,
dG = VdP ------------- (1)
On going from state 1, with free energy G1, to state 2, with free energy G2, by a reversible path, the change in free energy (G2-G1) is the sum over the path, or integral, of all the dG values along the path, as long as the process is reversible.
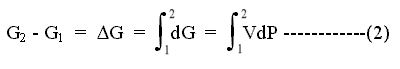
Since P is related to V by:
PV = nRT
where n is the amount of gas in moles, and R is the gas constant. From the rules of integration:
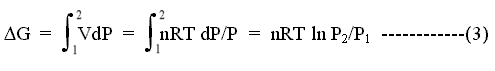
We now define our initial state as a standard state in which the gas was at a pressure of 1 atm. Then
G2 = Go + nRT ln P2 --------------------------- (4)
where Go is the standard state free energy of the gas.
In a mixture of ideal gases, each gas has a partial pressure which is the pressure which the gas would have if it alone occupied the volume. The partial pressure is thus equivalent to the concentration of the gas. For gases A, B, C, D the partial free energy due to each gas (G#A, G#B, G#C, G#D) is related to the partial pressure of the gas by equation 4:
G#A = G#oA + nART ln PA, G#B = G#oB + nBRT ln PB, etc.
Let us now consider an ideal gas in equilibrium with the gas in solution. The concentration of the gas in solution (assuming an ideal solution) will be proportional to the pressure of the gas, so that an equivalent set of equations must exist relating the partial free energy of the gas in solution to its concentration, X.
The partial free energy per mole of a component is its chemical potential, so we may write:
GA#soln = nAµA, GB#soln = nBµB, etc.
and, dividing by nA, nB, etc.
GA#soln = µA = µoA
+ RT ln XA,
nA
GB#soln = µB = µoB
+ RT ln XB, etc. --------(5)
nB
where XA, XB, etc., are concentrations of the gases in solution.
Note that by dividing G# by n, we have changed the units from free energy (measured in joules, J, or calories,) to free-energy per mole (J.mol-1, or cal.mol-1). This is a more useful parameter for comparing energy terms for different chemical processes, because we don't have to worry about the size of the system when comparing the work potential available from different reactions.
For non-ideal solutions, the relation between concentration and pressure of a gas in equilibrium with the solution is affected by interaction between molecules in the solution. This leads to a deviation from the constant proportionality between pressure and concentration, so that equations 5 do not apply exactly. This deviation is compensated by considering activities (aA, aB, etc.) instead of concentrations. The activity is related to concentration by an activity coefficient (g), so that
a = gX
Then
µA = µoA + RT ln aA -------- (6)
= µoA + RT ln XAgA
In dilute solution, the activity coefficient approaches unity so that the behavior of the molecules approaches the ideal, and activities approximate to concentrations. As we will see below, when considering the energy changes in reactions, activity terms occur in ratios, and activity coefficients tend to cancel. (Note that in equation 6, a term of value unity, corresponding to the activity in the standard state, is ommitted by convention from the logarithmic term.)
A + B <===> C + D
At equilibrium, the reactants and products have activities [A], [B], [C], [D], so that
K = [C].[D]
[A].[B]
Let us assume that the reaction is poised away from equilibrium by the application of work at a new poise with activities [A'], [B'], [C'], [D']. The change in partial free energy per mole (chemical potential) for each reactant as the reaction proceeds reversibly back to the true equilibrium is given by:
![]() µA = µ'A
- µA = µoA + RT ln [A'] -
µoA - RT ln [A] = RT ln [A']/[A]
µA = µ'A
- µA = µoA + RT ln [A'] -
µoA - RT ln [A] = RT ln [A']/[A]
![]() µB = µ'B
- µB = µoB + RT ln [B'] -
µoB - RT ln [B] = RT ln [B']/[B]
µB = µ'B
- µB = µoB + RT ln [B'] -
µoB - RT ln [B] = RT ln [B']/[B]
![]() µC = µ'C
- µC = µoC + RT ln [C'] -
µoC - RT ln [C] = RT ln [C']/[C]
µC = µ'C
- µC = µoC + RT ln [C'] -
µoC - RT ln [C] = RT ln [C']/[C]
![]() µD = µ'D
- µD = µoD + RT ln [D'] -
µoD - RT ln [D] = RT ln [D']/[D]
µD = µ'D
- µD = µoD + RT ln [D'] -
µoD - RT ln [D] = RT ln [D']/[D]
Then, adopting the convention that changes in the concentrations of the products are positive
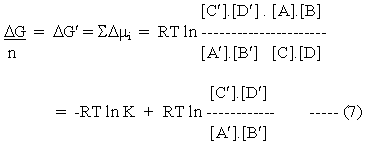
Let us assume that the activities of the reactants poised away from
true equilibrium were those of the standard states, [Ao], [Bo],
[Co], [Do]. By definition, these have the value 1,
so that the second term on the right of equation 7 becomes zero. Then the
free energy change for a reaction going from the standard state back to
equilibrium (![]() Go) is
given by
Go) is
given by
![]() Go = -RT ln K -------
(7a)
Go = -RT ln K -------
(7a)
and
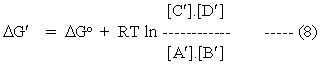
In order to consider reactions in which the stoichiometric coefficients are not equimolar, we simply extend the relationships of equations 7 and 8 to the more general case.
aA + bB <===> cC + dD
and
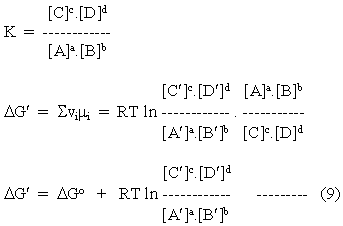
Equation 9 is generally applicable to all chemical reactions, and we
shall use it in describing biochemical processes. Note that ![]() G'
has units of J.mol-1,- it is an intensity factor, and must be
multiplied by n, the number of moles of reactant undergoing the chnage
in state represented by the reaction, to get the free energy change.
G'
has units of J.mol-1,- it is an intensity factor, and must be
multiplied by n, the number of moles of reactant undergoing the chnage
in state represented by the reaction, to get the free energy change.
![]() G = n
G = n ![]() G'
G'
![]() G',
G', ![]() Go,
and
Go,
and ![]() Go' are more
convenient parameters for comparing the free energies available from chemical
reaction, because they are normalized so as to avoid the need to take account
of the size of the system.
Go' are more
convenient parameters for comparing the free energies available from chemical
reaction, because they are normalized so as to avoid the need to take account
of the size of the system.