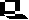
As we saw in our discussion of oxidation levels, one of the unique characteristics of carbon is that it has nine stable oxidation states. It should not be surprising that organic chemists have developed reagents that allow them to alter these oxidation levels. This topic presents a survey of some of those reagents.
We have already seen several examples of such reagents in our discussion of the oxidation of alcohols. They are repeated here for the sake of completeness.
This reagent is prepared by mixing sodium or potassium dichromate with sulfuric acid as shown in Equation 1.
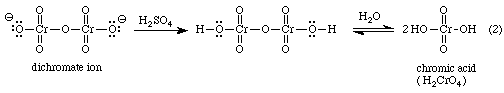
It is used to oxidize secondary alcohols to ketones:
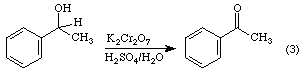
It may also be used to oxidize primary alcohols to carboxylic acids. As Equation 3 indicates, the alcohol is initially oxidized to an aldehyde. Under the reaction conditions, a molecule of water adds to the carbonyl group to form a hydrate which is subsequently oxidized to the carboxylic acid.
In order to prevent aldehydes from further oxidation, it is necessary to avoid the addition of water to the carbonyl group. PCC was developed as a non-aqueous alternative to chromic acid. Using this reagent, 2-phenylethanol may be oxidized to phenylacetaldehyde without subsequent oxidation to phenylacetic acid:
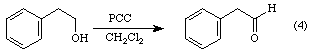
These reagents are used to convert alkenes into the corresponding 1,2-diols (glycols) by a process called syn hydroxylation. Equation 5 illustrates the process for the reaction of 1,2-dimethylcyclohexene with a dilute solution of potassium permanganate.
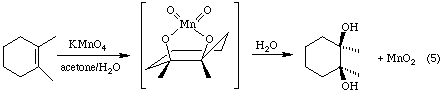
The reaction is thought to involve the formation of an intermediate cyclic permanganate ester which is readily hydrolysed under the reaction conditions to yield the 1,2-diol. A cyclic osmate ester is generated with OsO4.
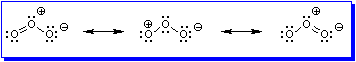
Ozone, O3, is an allotrope of oxygen. It is a highly reactive molecule that is generated by passing a stream of dioxygen over a high voltage electric discharge. (It is possible to smell ozone in the atmosphere after a lightning storm if the lightning has struck nearby.) It is not possible to draw a single structure for O3 in which each oxygen atom has a filled valence shell and is, at the same time, uncharged. Rather, resonance theory describes the structure of this compound as a hybrid of the three resonance contributors shown in Figure 1.
In a process called ozonolysis, an alkene is treated with ozone to produce intermediates called ozonides, which are reduced directly, generally with zinc metal in acetic acid, to yield aldehydes or ketones, depending on the substituents attached to the double bond of the initial alkene. Equations 6 -8 provide three specific examples.
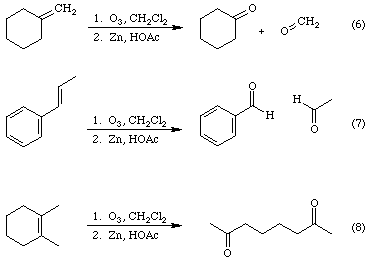
Note that an aromatic ring is resistant to ozone.
The value of ozonolysis lies in the structural insight it affords a chemist who is trying to determine the identity of an unknown compound. Figure 2 illustrates this idea.
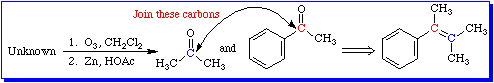
The unknown is degraded into smaller, simpler molecules that are more readily identified. Once identified, these fragments are then mentally reconnected by joining the carbonyl carbons together to create an alkene.
In our discussion of the oxidation of alcohols, we classified this process as a 1,2-elimination of the "elements of" dihydrogen. The reverse process, the 1,2-addition of the "elements of" dihydrogen to a multiple bond, constitutes a reduction. The reagents used for the 1,2-addition of the "elements of" dihydrogen to a multiple bond depend upon the nature of the multiple bond. For homonuclear multiple bonds, i.e. alkenes and alkynes, the most common method is called catalytic hydrogenation: a solution of the alkene or alkyne is mixed with dihydrogen gas in the presence of a catalytic quantity of a transition metal. For heteronuclear multiple bonds; aldehydes, ketones, nitriles, esters, etc., addition of the "elements of" dihydrogen is genarally accomplished in two steps, addition of hydride ion, :H-, followed by addition of H+. Since the addition of hydride ion is rate determining, these reductions are called hydride ion reductions. We'll take a look at catalytic hydrogenation first.
The catalyst most commonly used to reduce carbon-carbon multiple bonds consists of platinum metal dispersed over the surface of finely divided charcoal (Pt/C). Palladium and rhodium are also used (Pd/C and Rh/C). These reagents are available commercially. The catalyst is mixed with a solution of the alkene or alkyne dissolved in an inert solvent such as ethanol or diethyl ether. Since the reaction mixture is heterogeneous, it is important that the catalyst be dispersed over a large surface area in order to insure adequate contact between the reactants and the catalyst. The charcoal provides the required surface area.
The mechanistic details of catalytic hydrogenations are uncertain because of the difficulties associated with studying heterogeneous reactions. Figure 3 animates the generally accepted view of the process using 2-butyne as an example.

Adsorption onto the catalytic surface brings the reactants into proximity. It also weakens the H-H and the C-C bonds, increasing the reactivity of the reactants. The details of the transfer of the hydrogen atoms from the platinum to the alkyne are uncertain. However, as the animation indicates, both hydrogen atoms add to the same side of the pi bond, leading to the formation of cis-2-butene. In other words, catalytic hydrogenation of alkenes and alkynes involves the syn addition of the "elements of" dihydrogen to the multiple bond. Equation 9 illustrates the syn addition of dihydrogen to (E)-3-chloro-2-phenyl-2-butene. Since the addition occurs to the top and bottom faces of the pi bond with equal probability, the reaction produces a racemic mixture of the two stereoisomers shown.
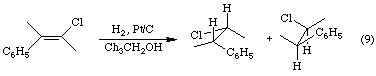
As reaction 10 indicates, catalytic hydrogenations are not restricted to alkenes and alkynes. Compounds containing multiple bonds between carbon and a heteroatom such as oxygen or nitrogen may also be reduced catalytically.
While the outcome depicted in reaction 10 may be desireable, it would be nice to have a method to hydrogenate heteronuclear multiple bonds while leaving carbon-carbon multiple bonds untouched. Such a method exits. It takes advantage of the fact that, unlike C-C multiple bonds, which are relatively non-polar, heteronuclear multiple bonds have a permanent bond dipole; the carbon is electron deficient while the heteroatom is electron rich. This means that the carbon atom of a heteronuclear multiple bond is inherently reactive toward negatively charged reagents, while the heteroatom is inherently reactive towards positively charged reagents. Figure 4 demonstrates this reality for negatively and positively charged hydrogen atoms.
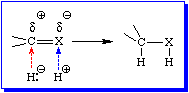
The net result of the addition of :H- and H+ to the multiple bond is the 1,2-addition of the "elements of" dihydrogen. Since it is not possible to have significant concentrations of :H- and H+ in the same flask at the same time (why?), hydrogenation of heteronuclear multiple bonds is normally a 2-step process; hydride ion adds to the carbon atom in the first step, while a proton adds to the heteroatom in the second. The remainder of this topic will consider different reagents that act as a source of hydride ion.
Reagents that act as hydride ion donors all share one structural feature: They all contain at least one hydrogen atom that is bonded to another atom which is less electronegative than hydrogen. The greater the difference in electronegativity, the more reactive the reagent will be as a hydride donor. The most reactive source of hydride ion is lithium aluminum hydride, LiAlH4. This material is a grey solid that reacts violently with protic solvents. Most commonly it is used as a suspension in a dry, inert solvent such as diethyl ether or THF. A solution of the compound to be reduced is added to this suspension and stirred vigorously until analysis indicates that all of the starting material has reacted. At this point the mixture is acidified by the careful addition of aqueous acid. Figure 5 illustrates these two steps for the reduction of acetophenone.
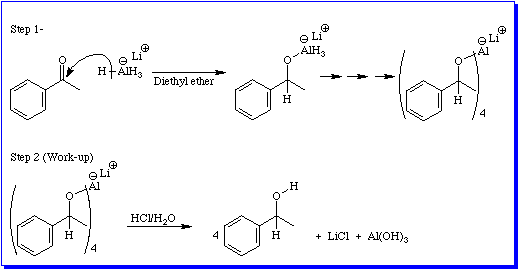
Note that all four hydrogen atoms attached to the aluminum in LiAlH4 are active; one mole of LiAlH4 will reduce four moles of the ketone.
LiAlH4 is so reactive that it will reduce almost any type of heteronuclear multiple bond. It will even reduce carboxylic acids and esters to the corresponding primary alcohols as indicated in reactions 11 and 12, and it reduces amides to amines as shown in Equation 13.
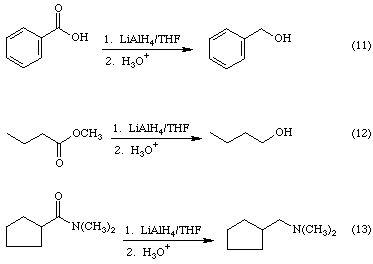
Clearly these reactions are more complicated than the mechanism shown in Figure 5 would suggest. Elimination of water as well as reduction must be involved.
Before we consider less reactive hydride donors, let's revisit the reaction of the unsaturated ketone we considered in Equation 10. Figure 6 compares the catalytic hydrogenation of pent-4-en-2-one with its reduction by LiAlH4.
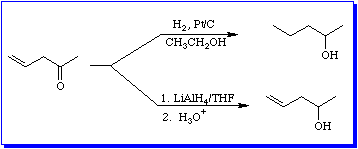
Because simple, i.e. non-conjugated, double bonds are non-polar, they are non-reactive towards nucleophilic reagents.
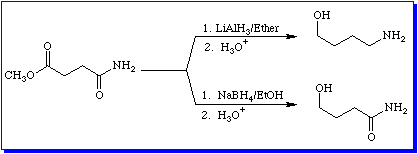
Now let's consider another aspect of reactivity. For compounds that belong to the carboxylic acid family, in particular carboxylic acids, esters, and amides, the oxidation level of the carboxyl carbon is +3. As you can see from the reactions in Figure 7, the oxidation level of the carboxyl carbon decreases to -1 when the carboxyl group is reduced to a primary alcohol. The question then becomes, "Can you stop the reduction at an intemediate oxidation level?" The answer is yes. Equation 14 shows how.
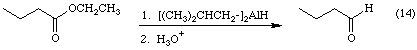
Here the ester is reduced to an aldehyde. The oxidation level of the carbonyl carbon decreases from +3 to +1. The trick is to use a sterically hindered reducing agent that has only one active hydrogen. In this case the reagent is called diisobutyl aluminum hydride, sometimes abbreviated DIBAL. By using 1 equivalent of DIBAL at low temperatures it is possible to reduce the ester to the corresponding aldehyde without further reduction of the aldehyde to the primary alcohol. Use of more than 1 equivalent will lead to reduction of the aldehyde.
Finally, Table 1 summarizes the reactivities of the various reducing reagents we have considered in this topic.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|