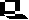
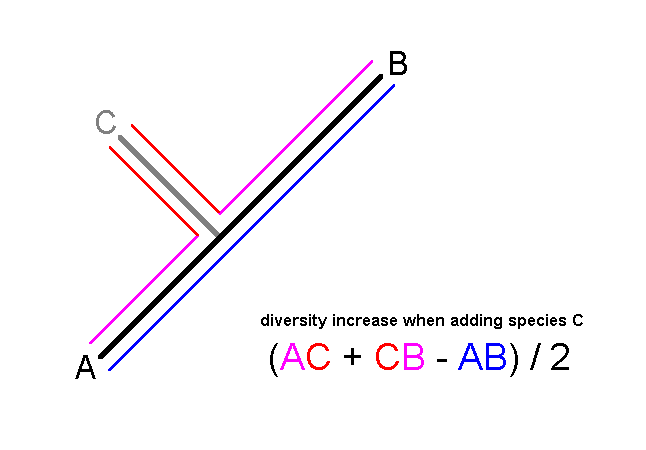
To illustrate how classifications may be used to predict diversity value
by estimating richness at the level of the genes or characters, the example
below shows a classification for some African species of milkweed butterflies
(family Nymphalidae). The branching pattern of the classification is derived
by analysis of 217 morphological and chemical characters, and the branch
lengths are scaled by the number of character changes found within this
sample (shown as vertical ticks). Considering all combinations of three
species, the most diverse set of three species is niavius, echeria
and damocles, because these three have the longest total branch
lengths with the largest numbers of character differences between them
(shown in black) (below):

To illustrate how using this approach can effect the relative values of different faunas or floras, consider first one of the most popular measures of diversity, species richness. The example below shows counts of the numbers of species of sibiricus-group bumble bees among equal-area grid cells (below):
 |
Link to image showing results of species richness measure. |
|
|
Link to image showing results of phylogenetic diversity measure. |
| The �clock�
(anagenetic) model
assumes that changes occur at random and are subject to little constraint by selection. Consequently, in effect, changes accumulate more or less in proportion to the time elapsed along the branches. |
|
Result:
if sample data are unavailable or are expected to be biased and unrepresentative, all lineages are scaled to a common length. |
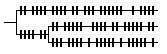 |
 |
|
|
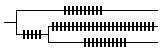
The sample (empirical) model assumes that the distribution of changes in a small sample of variation is representative of the great majority of unsampled variation. The pattern of changes is usually expected to be intermediate between those from the other two models. |
|
Result: if sample data are available and are expected to be representative, all branches are scaled in proportion to sample changes. |
 |
 |
|
|
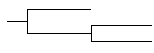
The saltatory (cladogenetic) model assumes that most changes are associated with speciation or divergence events, for example if strong selection constraints are relaxed at these times. Although the numbers of changes associated with each branching event may differ, in effect changes accumulate more or less in proportion to the number of branching events (including those to extinct branches). |
|
Result: if sample data are unavailable or are expected to be biased and unrepresentative, all branches are scaled to unit length. |
 |
 |
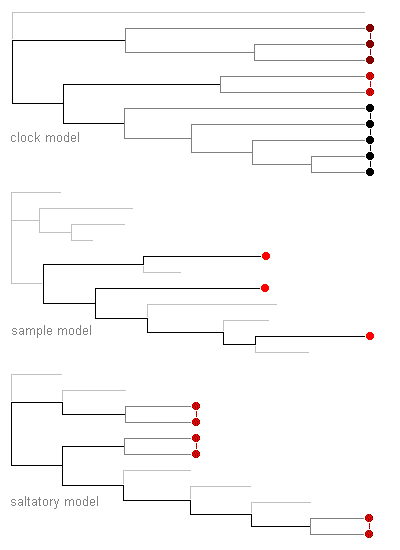
Most disagreement is now centred on the choice of special evolutionary
model (ref 2). The sample model is the
easiest to use, and its consequences in this example show that it may given
less equivocal answers concerning which species represent the greatest
relative diversity.
However, the apparent advantages of the sample model are only real
if the sample truly represents the overall pattern of variation. For example,
if a sample of unconstrained genetic data were available that behaved as
though following the clock model, whereas the value being sought for expressed
characters was under strong stabilising selection and distributed as though
it followed the saltatory model, then using the sample model could introduce
a severe bias into the measure. Consequently, any apparent increase in
resolution arising from using information from the sample in this way could
actually be misleading.