Die erste Frage ist am einfachsten zu beantworten. Das vergleichende Studium bestimmter Nukleinsäurefraktionen (rRNS) führte
| zur Einteilung der Prokaryoten in Eu- und Archaebakterien, | |
| zum Nachweis von Homologien zwischen der rRNS aus Chloroplasten und Cyanophyceen. Dies erwies sich als sicherste Stütze der Endosymbiontenhypothese, | |
| zur Eingrenzung des Zeitpunkts der Abzweigung einzelner Stammeslinien
im Pflanzenreich (H. HORI et al., 1985). |
|
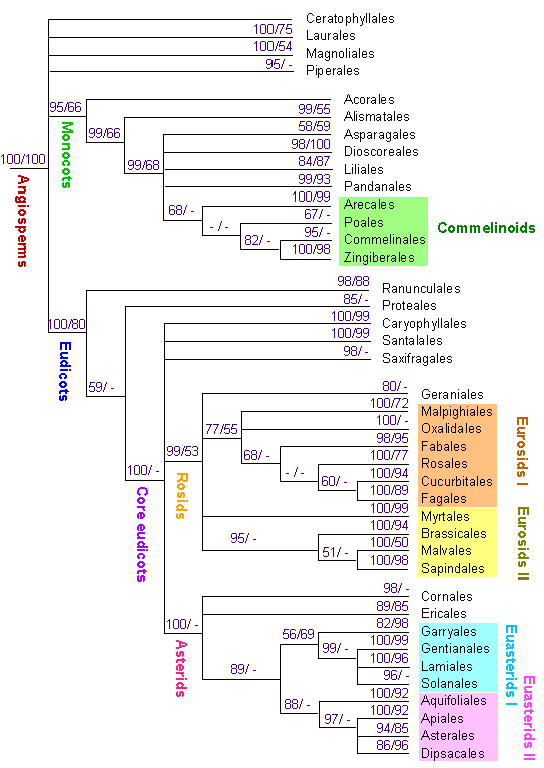
| und schließlich zur Klärung der phylogenetischen Beziehungen der wichtigsten Gruppen der grünen Pflanzen, der Landpflanzen, der Samenpflanzen und der Angiospermen. Der Analyse lag der Vergleich der Sequenzen von drei Genen (rbcL, atpB, and 18S rDNA) zugrunde (KÄLLERSJÖ, M., J. S. FARRIS, M. W. CHASE, B. BREMER, M. F. FAY, C.J. HUMPHRIES, G. PETERSEN, O. SEDERG & K. BREMER 1998. Simultaneous parsimony jackknife analysis of 2538 rbcL DNA sequences reveals support for major clades of green plants, land plants, seed plants and flowering plants. Pl. Syst. Evol. 213, 259-287), Für die Angiospermen wurde von der Angiosperm Phylogeny Group das folgende phylogenetische Schema erarbeitet, |
Die elektronische Version des Dendrogramms (veröffentlicht in den Annals of the Missouri Botanical Garden 85: 531-553.(1998) wurde von R. BERGMANN & P. v. SENGBUSCH entwickelt. Es wurde in eine interaktive Datei umgesetzt, Die Links führen zu den Familienlisten der APG Klassifikation.
Einen Überblick über die aktuell vorliegenden Nukleotidsequenzen
können inzwischen nur noch über gezielte Suche in Datenbanken gewonnen werden.
Einen Einstieg hierzu bieten die Angebote des Europäischen Laboratoriums für
Molekularbiologie,
Anders sieht es bei den Proteinen aus. Nachdem F. SANGER (MRC Laboratory of Molecular Biology, Cambridge/Engl.) Mitte der fünfziger Jahre Methoden zur Sequenzierung ausgearbeitet hatte, glaubte man, sie für das Studium phylogenetischer Zusammenhänge nutzen zu können. Ausgangspunkt dieser Überlegung war die Feststellung, daß Proteine primäre Genprodukte seien. Mit anderen Worten: Es seien unveränderte Translationsprodukte. Wenn man von den inzwischen bekannten Einschränkungen (Processing von RNS, Posttranslationsmodifikation usw.) absieht, kann man diese Aussage als Arbeitshypothese stehen lassen.
Morphologische Merkmale unterliegen, wie schon ausführlich dargestellt, der Selektion. Sie können durch vielerlei Einflüsse abgeändert werden und bringen Systematiker dadurch in Schwierigkeiten, weil sie nicht mehr die Stammesgeschichte allein widerspiegeln. Erschwert wird die Situation zudem noch durch Analogie und Konvergenz.
Durch das Studium von Proteinen hoffte man, diese Probleme umgehen zu können. Man glaubte, von einer nicht-darwinistischen Evolution ausgehen zu können, d.h., einer Anreicherung von Mutationen (hier Veränderungen einzelner Aminosäuren in der Polypeptidkette) als Funktion der Zeit. Man sprach daher auch von einer molecular clock oder den "Uhren der Evolution".
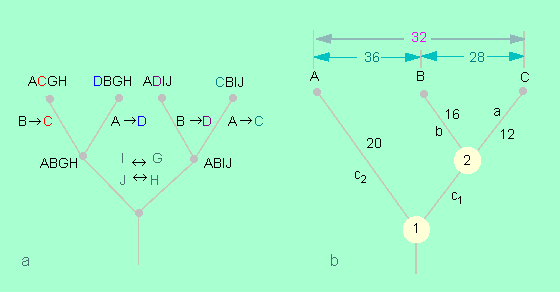
Aufstellung von Stammbäumen aufgrund von Vergleichen verschiedener Aminosäuresequenzen des gleichen Proteins bei verschiedenen Arten. a: Zuordnung von Sequenzen aufgrund der geringstmöglichen Zahl von Aminosäureveränderungen. Man kann auf diese Weise zurückextrapolieren und die wahrscheinlichsten gemeinsamen Sequenzen ermitteln. b: Aussagen aufgrund der Zahl der Austausche. Hieraus läßt sich der Grad der Verwandtschaft von Arten ablesen. Wenn zwischen den Arten A und C 32 Austausche liegen, zwischen B und A 36 und zwischen B und C 28, so läßt sich extrapolieren, wie viele davon auf die einzelnen Linien entfallen, nachdem sich die beiden Arten jeweils getrennt voneinander entwickelt haben (Nach M. O. DAYHOFF, 1972).
Es gibt einige Proteine (z.B. das Cytochrom c, welches einen Schritt in der Atmungskette katalysiert), die relativ leicht aus Zellen der verschiedensten Organismen isolierbar sind. Die Funktion des Cytochroms c ist in allen Organismen die gleiche. Die Aminosäurezusammensetzung seiner Polypeptidkette variiert. Je höher der Verwandtschaftsgrad zwischen zwei Taxa ist, desto größer sind die Gemeinsamkeiten. Die Polypeptidkette besteht bei allen Tieren aus 104, bei allen untersuchten höheren Pflanzen aus 112 Aminosäureresten. (D. BOULTER, University of Durham).
Wählt man Cytochrom-c-Daten von Arten aus verschiedenen Organismenreichen oder Stämmen, zeigt sich, daß die obengenannte Annahme zutreffend ist. 1972 lagen genügend Analysen über das pflanzliche Cytochrom c vor, um einen ersten Versuch zur Rekonstruktion phylogenetischer Zusammenhänge zu wagen. Bereits dabei zeigte sich, daß diese Daten nicht mit den Verwandtschaftsbeziehungen zwischen Pflanzengruppen übereinstimmten, die man aufgrund traditioneller Methoden erzielt hatte. Statt in Form eines Baumes, mußten die Cytochrom-c-Daten in Form eines "Strauchs" dargestellt werden.
BOULTER und Mitarbeiter versuchten die Kontroverse zu lösen, indem sie anstelle des Cytochroms c andere - bei Pflanzen weitverbreitete - Proteine analysierten, z.B. Phycocyanin und einige Ferredoxine. Aber auch hier zeigte sich, daß proteinchemische und allgemein akzeptierte morphologische Daten nicht immer in Einklang zu bringen waren. Die jeweils homologen Proteine aus nah verwandten Arten, z.B. Cytochrom c aus Brassica oleracea und Brassica napus, oder Plastocyanin aus zwei Heracleum-Arten, oder Ferredoxine aus mehreren Equisetum-Arten, erwiesen sich als gleich oder fast gleich. Hier werden Verwandtschaftsverhältnisse also so widergespiegelt, wie man es erwarten würde. Auch beim Vergleich von verwandten Gattungen schneiden proteinchemische Vergleiche ganz gut ab. So ähnelt z.B. das Ferredoxin von Triticum dem von Aegilops, und das Plastocyanin von Solanum tuberosum dem von Lycopersicon esculentum. Doch auf Familienebene treten meist Diskrepanzen auf. Zwar sind die Ähnlichkeiten innerhalb der Familien so wie erwartet, aber die Beziehungen der Familien zueinander bleiben vielfach unerklärbar.
Woran liegt das? Offensichtlich gibt es auf Proteinebene einen hohen Grad an Polymorphismus. Wie schon des öfteren erwähnt, sind Genduplikationen und Polyploidisierungen im Verlauf der Angiospermenentwicklung häufig anzutreffen. Welches Allel und welches der Gene im einzelnen dabei zum Zuge kommt, ist offen. Schon das erschwert die Interpretation proteinchemischer Daten.
Ein zweiter, entscheidender Punkt: Es wurde nach und nach deutlich, daß Proteine in ihrer Zusammensetzung ebenso wie alle übrigen Strukturen und Funktionen einer Selektion unterworfen sind. Nur, der Selektionsdruck, der auf ein Protein ausgeübt wird, unterscheidet sich von dem, dem die Individuen ausgesetzt sind. Die Folge ist eine unterschiedliche Evolutionsgeschwindigkeit der einzelnen Proteine einerseits, der Organismen andererseits. Das beste Beispiel hierfür muß wieder dem Tierreich entnommen werden: Die Variation der Albumine (Blutplasmaproteine) zwischen Froscharten ist gleich groß wie zwischen Säugerarten. Trotz gleichbleibender Änderungsrate der Albumine erfolgte die Evolution körperlicher Merkmale bei den Säugern sehr rasch, bei den Fröschen tat sich dagegen fast nichts.
Diese Einwände machen deutlich, daß man durch die Analyse von Proteinen zwar etwas über Gene bzw. Allele lernen kann, daß aber einzelne exprimierte Gene nur wenig über das Genom und dessen Organisation und Expression aussagen. Zum Verständnis der Evolution und Entwicklung von Organismen ist dies aber auch mit entscheidend.
Sequenzanalysen von Proteinen sind außerordentlich zeitraubend und kostspielig. Das allein rechtfertigt die Ausarbeitung von Näherungsverfahren, mit deren Hilfe man die Identität oder Unterschiede zwischen beliebig vielen Proben erfassen kann. Zwei Verfahren haben sich als besonders günstig erwiesen: die Gelelektrophorese (einschließlich einer Variante: der Isoelektrischen Fokussierung) und die Serologie. Je nach experimentellem Ansatz lassen sich gelelektrophoretisch das Molekulargewicht, die Anzahl der Ladungen und/oder die Aktivität bestimmter Enzyme bestimmen. Dadurch ergeben sich Lösungsansätze zu der vorhin aufgeworfenen Problematik: Wie unterscheidet man Proteine als Produkte verschiedener Gene oder Allele? Die Ergebnisse derartiger Untersuchungen wurden bereits mehrfach ausführlich diskutiert.
Man kann mit Hilfe dieser Technik Allo- und Isoenzymmuster analysieren. Das Studium der Isoenzyme hat maßgeblich zum Fortschritt der Entwicklungsphysiologie beigetragen, das Studium der Alloenzyme zum Fortschritt der Evolutionsforschung und zur Analyse von Populationsstrukturen und Verwandtschaftsverhältnissen. Eine herausragende Rolle spielen dabei die Untersuchungen an der Ribulose-1,5-Bisphosphatcarboxylase.
Während man durch elektrophoretische Analyse vornehmlich Ladungsunterschiede in Proteinen erfaßt, lassen sich mit serologischen Verfahren unterschiedliche Oberflächenstrukturen von Proteinen nachweisen. Ein Antikörper gegen Protein A wird mit einem Protein B nur dann reagieren (eine Kreuzreaktion zeigen), wenn beide gemeinsame antigene Determinanten an ihren Oberflächen exponiert haben. Je größer die Gemeinsamkeiten, desto deutlicher fällt die Kreuzreaktion aus.
In einer Polypeptidkette mit über 100 Aminosäureresten genügt bereits ein veränderter Aminosäurerest, um einen serologischen Unterschied sichtbar werden zu lassen. In kleinen monomeren Proteinen beeinflußt praktisch jede Aminosäureveränderung (Mutation) die Oberfläche und wird daher serologisch erkannt. In großen multimeren Proteinen ist das nicht immer der Fall.
Der Grad serologischer Verwandtschaft ist von verschiedenen Autoren zum Studium phylogenetischer Zusammenhänge von Taxa unterschiedlicher Kategorien herangezogen worden. Als Proteine eignen sich vorzugsweise die aus Samen isolierten Speicherproteine sowie eine Anzahl von Enzymen. Auch die so gewonnenen Ergebnisse zeigen, wie wichtig es ist, Merkmale zu studieren, die sich von den übrigen Merkmalssätzen unterscheiden. Verwandtschaft zwischen Arten, Gattungen und Familien ist nachweisbar, die Daten können zu Dendrogrammen zusammengefaßt werden. Vielfach, doch nicht immer, stimmen serologische Verwandtschaft und morphologisch nachgewiesene Verwandtschaft miteinander überein; gelegentlich gaben die serologischen Befunde den Ausschlag, fragliche Verhältnisse zu klären.
© Peter v. Sengbusch - b-online@botanik.uni-hamburg.de