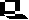
Peptides - especially polypeptides or proteins - play an outstanding part in every cell. They act as biological catalysts (= enzymes), take part in the regulation of the cell's metabolism and in the interaction between cells and are required for the generation of specific structures. They are linear chains consisting of a sequence of amino acids linked exclusively by peptide bonds.
|
|
This type of linkage causes a polarity, since - independent of the chain length - a free amino group terminates one end of the chain, the N-terminus, while the other is terminated by a free carboxyl group (C-terminus). Amino acid sequences are written from N- to C-terminus, the direction in which protein synthesis proceeds. The exact sequence of amino acids (also called the protein's primary structure) is determined by the nucleotide sequence of the accompanying nucleic acids and characterizes a certain, specifically acting protein molecule.
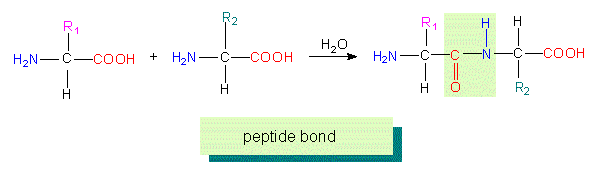
Protein synthesis is a costly, complex process in which the genetic information stored in the DNA is finally translated into the amino acid sequence of the protein. The mode of synthesis may be different in short peptides. In many cases, the amino acids are linked to short chains by the respective enzymes. This mode allows also the use of rare amino acids that do not belong to the 20 amino acid standard. But there are cases, too, where a normal amino acid within a protein is modified enzymatically later on. An example is the change of proline to hydroxyproline.
For the provisional characterization of a protein it is necessary to know
![]() how
many amino acids the polypeptide chain contains,
how
many amino acids the polypeptide chain contains,
![]() which
amino acids these are and what sequence they have.
which
amino acids these are and what sequence they have.
The primary structures of numerous proteins are known, examples
are the coat protein of tobacco
mosaic virus (TMV) or the cytochrome c from
yeast. The analyzed sequences (76679 entries on November,
28th, 1998) are available in data bases, being compiled by Swiss
scientist and published as the project SWISSPROT.
The data are also available from the data bases of the European Laboratory of Molecular Biology in Heidelberg or from the European Bioinformatics Institute in Hinxton/England. The data bases offer a wide range of search facilities and methods for mutual comparison of selected sequences:
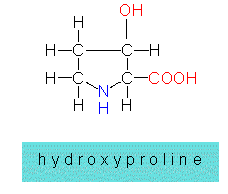
Though these data are very important, they are not sufficient. To understand the function of a protein it is necessary to know, how the polypeptide chain folds up. The chemical reactivity of the amino acid side chains cause an exactly determined, specific three-dimensional structure, the tertiary structure. While the primary structure depends exclusively on covalent bonds (peptide bonds), peptide bonds play only a minor part in the formation of the tertiary structure. The only bond that is important for the tertiary structure is the disulfide bond (disulfide bridge) that may form between the side chains of two cysteine residues.
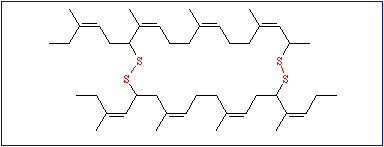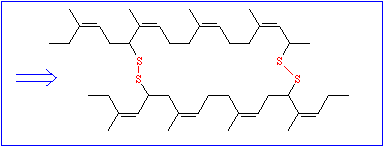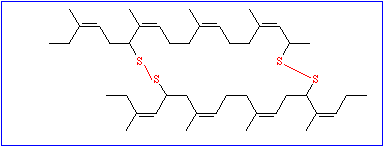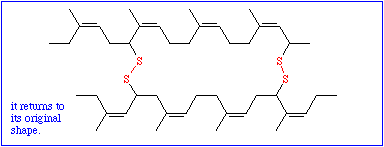
All other types of linkages belong to the weak interactions and are of the following kind:
![]() ionic
interactions,
ionic
interactions,
![]() hydrogen
bonds,
hydrogen
bonds,
![]() van-der-Waals-attractions.
van-der-Waals-attractions.
Regular foldings of the polypeptide chain, so-called secondary structures, may develop by hydrogen bond formation, producing the
|
|
alpha - helix: the most common, most stable and best-known type being an alpha - helix. Here, the hydrogen bonds form between neighbouring amino acids or such of the vicinity, so that the chain winds up to a helix. Stable, however, does not mean rigid. Due to the thermal movement all atoms within a molecule move against one another |
|
|
beta - pleated-sheets: Hydrogen bonds may also develop between the amino acids of different polypeptide chains that are arranged in an antiparallel or parallel way. This produces beta - pleated-sheets. |
.
The figure above shows the folding of one polypeptide chain only - Type of folding: Parallel beta-pleated-sheet. The original sheet forms only if more than two polypeptide chains (or parts of a long polypeptide chain) are arranged in parallel.
Anti-parallel beta-pleated-sheet: In this configuration, the polypeptide chains arrange in an antiparallel manner.
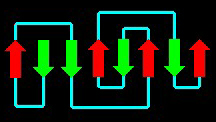
Anti-parallel and parallel beta-pleated-sheets (arrows)
in plastocyanin.
from: All-Beta Topologies -
Another impressive example of a protein with a high portion of beta-pleated-sheeets is concanavalin A
Secondary structures may be found in proteins though they are not necessarily present. Often, only small parts of a protein are involved in these structures. A tertiary structure, in contrast, forms always, although it is neither easily predictable how it is generated nor how the final molecular structure will look like.
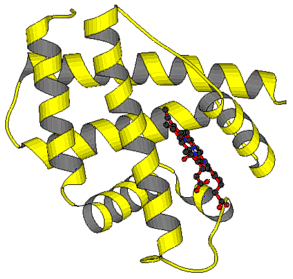
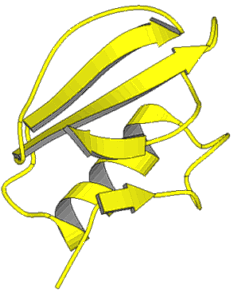
Examples of tertiary structures: To the left: : Leghemoglobin. The polypeptide chain folds mainly into alpha-helices. To the right: Chymotrypsin, a parallel beta-sheet (upper part of the picture), anti-parallel beta-sheet (at the bottom of the picture), alpha-helix (in the background). Tertiary structures can be displayed in various manners. Today, the information of the respective data base is used for the depiction of graphic models. They are subsequently processed with suitable programs Other examples. Bacterial chlorophyll, ConA, TMV (alternative representations: "backbone", "space-fill"), WGA
an interactive tutorial to demonstrate the special properties of Chime
 There
is only one method that gives definite information about tertiary
structures, X-ray analysis. The knowledge of the exact folding of
a protein is - especially for biologists - of eminent importance.
Only if the tertiary structure is known, explanations about the
actions of a certain molecule can be found. And only then it is
possible to say why a certain enzyme catalyzes this and no other
reaction, why its affinity is higher for one substrate than for
another or why a certain inhibitor
has an effect and how this effect is caused (competitive or
non-competitive inhibition). In other words: the development of a
tertiary structure transforms a linear information (the amino acid
sequence) into a three-dimensional structure. For the
understanding of biological processes - and generally for the
understanding of the mechanisms of life - the knowledge of
structures and their construction is imperative.
There
is only one method that gives definite information about tertiary
structures, X-ray analysis. The knowledge of the exact folding of
a protein is - especially for biologists - of eminent importance.
Only if the tertiary structure is known, explanations about the
actions of a certain molecule can be found. And only then it is
possible to say why a certain enzyme catalyzes this and no other
reaction, why its affinity is higher for one substrate than for
another or why a certain inhibitor
has an effect and how this effect is caused (competitive or
non-competitive inhibition). In other words: the development of a
tertiary structure transforms a linear information (the amino acid
sequence) into a three-dimensional structure. For the
understanding of biological processes - and generally for the
understanding of the mechanisms of life - the knowledge of
structures and their construction is imperative.
Picture to the right X-ray diagram of a proteine crystal (myoglobine), J. D. KENDREW, Cambridge. By evaluation of the diagram, the distribution of the electron density of the molecule is received, that has to be brought into line with a molecular model.
Many proteins are modified after biosynthesis. The original protein is activated by
|
|
splitting off of terminal parts, |
|
|
the chemical modification of single amino acids, for example by changing proline into hydroxyproline, by acetylation or phosphorylation or by |
|
|
association with further factors like coenzymes via weak interactions or covalent linkages with the polypeptide chain (apoenzymes). The actual enzymatic reaction does often take place at the coenzyme and not at the protein itself: coenzyme + apoenzyme = holoenzyme. |
Numerous functional proteins consist of several polypeptide chains. Such proteins are also called oligomer proteins. The single chains (subunits) may be the same or different. This type of organization is termed quaternary structure.
Proteins with quaternary structures may either have more than one enzymatic function or - and this is far more common - their enzymatic activity can be regulated by the substrate or other metabolites. The regulation principle is based on the tertiary structure of the subunits that is not completely rigid but can be distorted. In this way, the information that a substrate or a regulator has been bound to one subunit can be passed on via direct molecular contact to the other subunits so that their reactivity becomes modified (allosteric proteins; allosteric effects).
Although, for example, nearly the same enzymatically catalyzed reactions take place in a plant and an animal cell, the involved - homologous - enzymes are similar but never the same. They, too, have changed in the course of their organism's evolution. Such differences are therefore good markers for the reconstruction of phylogenetic lines.
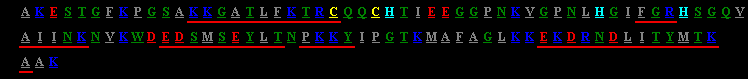
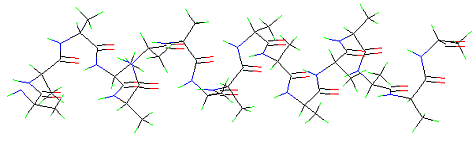
The amino acid sequence of cytochrome c from yeast (112 amino acid residues). The amino acids are given in the one-letter nomenclature, a colour code is used to label the reactive groups: grey: hydrophobic amino acids, green: hydrophilic amino acids, red: acidic amino acids (aspartic acid, glutamic acid), dark blue: basic amino acids (arginine, lysine), light blue: histidine, yellow: cysteine. Sequences underlined in red form alpha-helices.
The figure is taken as a screenshot from the project STING (Sequence To and withIN Graphics. The original URL is http://honiglab.cpmc.columbia.edu/STING/.
STING is a WWW tool for the simultaneous display of information about macromolecular structure (in STING's Graphics Frame) and sequence (in STING's Sequence Frame). Special attention is given to MacroMolecular INTERFACE analysis. (developed by: Goran Neshich's Lab. EMPBRAPA, Brasilia)
To use STING with your local files (PDB format), download STING and install it on your local server. To see 3D structures you need to install a CHIME Plugin. What you can do with STING:

Slide mouse over residue / nucleotide sequence in the Sequence Frame: Observe: residue / nucleotide number and its chain identifier on browser's (link reporting) lower border status field => STING's Status Frame!

Click on residue / nucleotide in the Sequence Frame: Observe: residue / nucleotide in CPK on Graphics Frame.

Click on Helix (red) or Extended (Blue) line below the sequence in the Sequence Frame: Observe: residue range modeled on ribbon on Graphics Frame.

Click with mouse on any atom in Graphics Frame (for PC users only): Observe: residue / nucleotide number and its chain identifier on browser's (link reporting) lower border status field => STING's Status Frame!

Click on any pre-defined CHIME scripts in STING's Control Frame: Observe: consequent actions in the Graphics Frame (see tutorial for detailed explanation of pre-defined CHIME scripts).

Click on any pertinent Data Base link in the STING's Control Frame: Observe: New browser being opened with pertinent data on PDB entry currently analyzed by STING.
Instant display of: GAPS in sequence, chain identifier, secondary structure elements type and range . This work was supported in part by grant DBI-9601463 from the US NSF. EMBRAPA / CENARGEN (Brasilia, Brazil) and specially Dr. Damares C. Monte and Dr. Afonso C. Valois are gratefully acknowledged for their support to BBNet/BBRC/BioInformatics Lab.
Finally, an example from the turorial
|
|